You know that feeling on your first day of A-Level chemistry when you find out everything they told you in GCSE was a lie? I felt something similar when I tried to find out which symptoms in ACS are actually helpful for ruling in/out the disease. It seems that few of the symptoms we are classically taught about are that useful, and the few that are reasonably good are not the ones we are taught about.
I first realised this whilst writing a new SBA for my app. I wanted to highlight what the criteria for suspecting ACS really were. This was based on an experience about a month ago where for about a week I was asked to see multiple patients/referrals from GPs for patients with chest pain that sounds like ‘classic’ ACS…until you clarify that the pain lasted seconds.
The SBA went like this:
A 53 year old man comes in with chest pain. When you assess him, the pain has subsided and he feels well. It was a central pain, occurring at rest and lasting about 5 minutes. There was radiation to the right, and there was no response to GTN. Which of these features makes the diagnosis of ACS most unlikely? A Duration of 5 minutes B Lack of response to GTN C Spontaneous resolution of pain combined with feeling well D Pain occurred at rest E Radiation to the rightI was basing this on the criteria given on the NHS Clinical Knowledge Summary, and the correct answer was meant to be A, as the pain needs to last at least 15 minutes to be suspicious of ACS. In agreement with the NICE guidance, the GTN response is meaningless. Pain has to occur at rest or with minimal exertion (or with less and less exertion for crescendo angina) to be ACS.

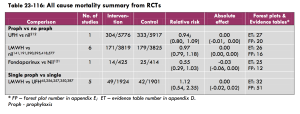
I was about to put this in the app when I decided to cross check it with SIGN, purely out of interest. When considering the question of what signs or symptoms are useful in ACS:
“A high quality systematic review of 21 studies examined the usefulness of 16 different clinical signs and symptoms in the diagnosis of acute coronary syndromes. Taken in isolation, no single sign or symptom was discriminatory.”
OK, so perhaps by using a cluster of symptoms we can rationally justify saying “I think this patient has ACS”, even if there is no single dealmaker/breaker symptom. I read the paper that SIGN’s conclusion was based on to check this.
To a certain extent, you could. Let’s look at the likelihood ratios for the various symptoms in MI. The likelihood ratio is the factor you multiply the pretest odds by to get the posttest odds. If a patient had central, crushing pain lasting 1 hour, by virtue of this history his probability of MI has gone up by 1.24 x 1.44 x 1.3 = 2.32. It would have been more significant if the pain radiated to the right than the combination of those factors. I will emphasise that. Radiation of the pain to the right confers a LR of 2.59. This compares to 1.45 for radiation of pain to the left.
It becomes even more counter everything-we-have-been-taught when you look at the LR for MI or unstable angina. Radiation of the pain to the right has a LR of 6.68 (admittedly with a CI of 2.95 to 15.2), which dwarfs all the other factors in the history that we classically use. Nothing else has an average LR of even 2 or more.
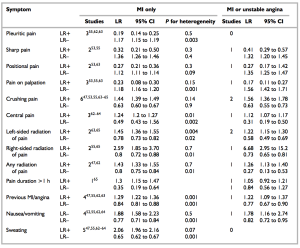
So it seems that perhaps we can use a combination of factors in the history, but we need quite a few of them. We should also respect right sided radiation as more worrying than left sided radiation.
(On a loosening of associations type trail of thought, RBBB is as common as LBBB in MI, and is associated with a worse in-hospital mortality. The SIGN guidance section 2.1 reflects this as well. It seems we’ve got our rights and lefts the wrong way round altogether in ACS. Also, LBBB does not usually mean MI, even if you have no old ECGs to compare to.)
There were some symptoms that were helpful at reducing the LR. Pleuritic chest pain had a LR of 0.19, and pain on palpation gave 0.23. Examination findings were also useful. I used the mnemonic MR HOPE to remind me about what examination findings matter in MI: MR = mitral regurgitation, HO = hypotension (LR 3.06) and PE = pulmonary edema (LR 2.08)/heart failure signs.
Simple enough. Let’s suppose we now have two patients with central crushing chest pain lasting 1 hour. This has a LR of 2.32. If a patient had a massive pre-test probability before this i.e. lots of CV risk factors, then this is potentially more significant than in a patient with no risk factors at all. This is why the risk factors are so important for ? ACS. It is the risk factors that determine your post-history ACS probability as much as the history of the presenting complaint itself. That said, it would be a brave doctor to discharge either patient without a 12 hour troponin.
12 hour troponins are the bane of hospital bed managers around the country. You have well patients occupying an acute medical bed for the next 10 hours whilst the GI bleeder is seen in the corridor. I wonder if this approach could be used when it comes to cardiac enzymes on a low risk patient. Myoglobin rises within 2-4 hours. It is really nonspecific, but highly sensitive. This means that it could be useful for excluding the diagnosis if negative, but not much use if positive. It’s like d-dimers. In fact, just like d-dimers, if it comes positive, you need to do the “real test” (12 hour troponin). Think how many beds not having a pretty well person hanging around for a 12 hour troponin could save.
(I know emedicine does not like this approach. However, if we were to restrict ourselves to using it after at least 4 hours, and on low risk patients, I think it could work. Need to do more reading into it though.)
Conclusions:
1. Myoglobins to be the new d-dimers. You read it here first.
2. Right sided radiation to be respected more. And history always interpreted in light of the patient’s risk factors (or “pre history of presenting complaint probability”)